Energy Minimization
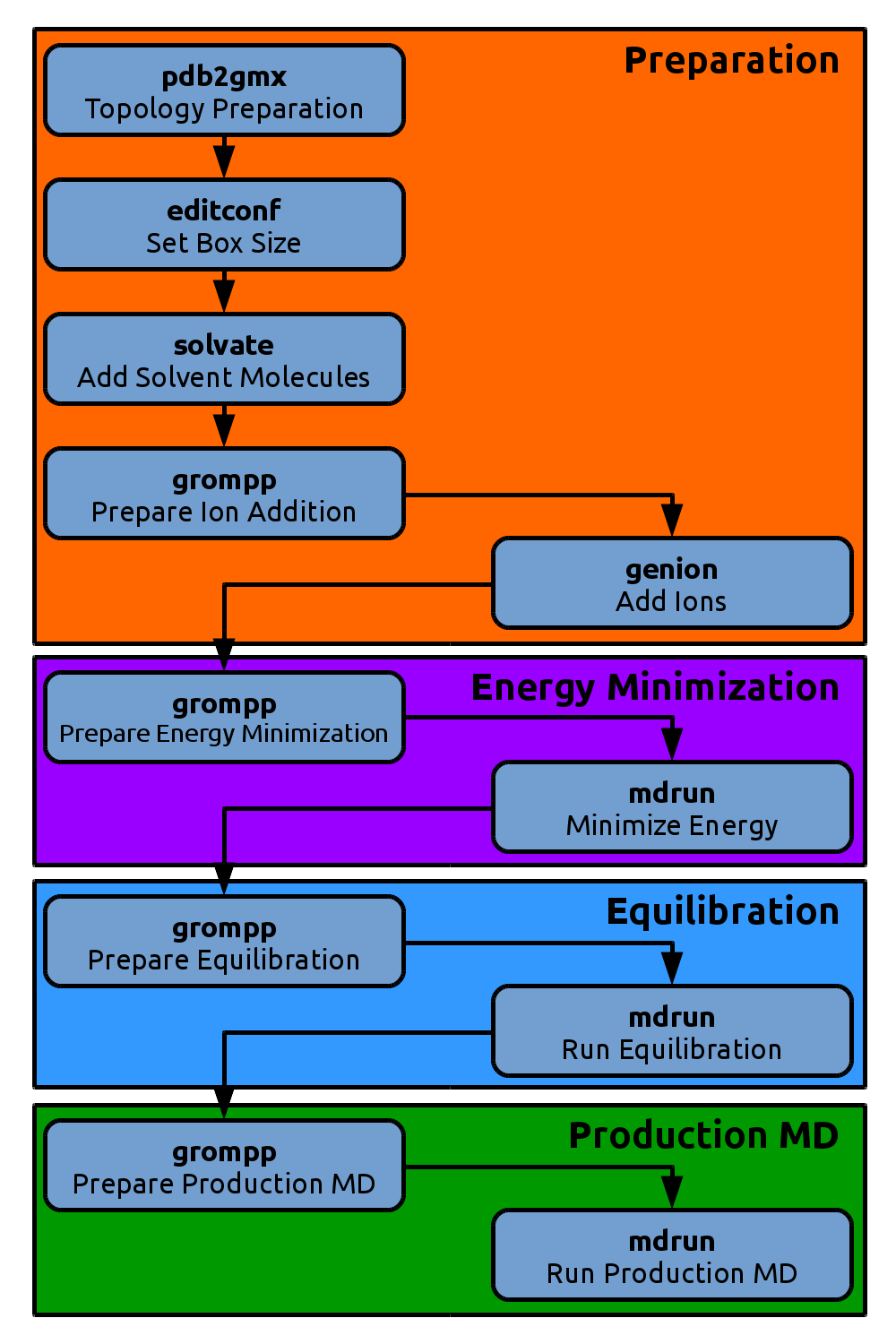
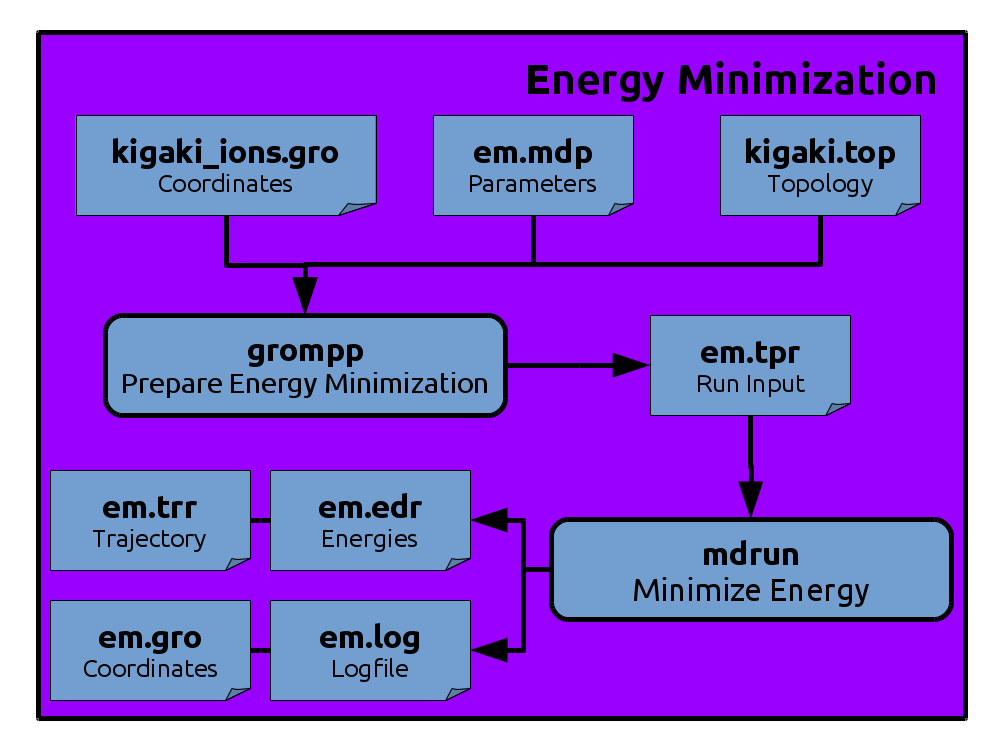
.tpr file with
grompp to combine the initial setup with the algorithms and
parameters from an .mdp file:
gmx grompp -f em.mdp -po em.out.mdp -c kigaki_ions.gro -p kigaki.top -o em.tpr
.tpr file that is the only file needed to
start our energy minimization.
Some new parameters are introduced in the file em.mdp.
Examine it and also have a look again at the
mdp section of the Gromacs Online Reference
to familiarize with all the parameters. We now use a steepest descent algorithm to locate the closest local minimum and set some convergence criteria. In addition we set the coordinate output frequency to every 10 steps with
nstxout = 10.
The energy minimization can be run with:
gmx mdrun -v -deffnm em >& em.out &
-v switch will make mdrun create verbose output and report the computation progress.
-deffnm defines a default filename prefix that is used for all in- and output files.
The corresponding file endings will be appended for the different in- and output files,
so they do not need to be specified each individually.
You will obtain a number of output files.
The file em.log contains detailed information on several energy terms throughout
the simulation and a performance analysis at the end.
The file em.gro contains the coordinates of the final configuration of the system.
The file em.trr contains coordinates (and optionally velocities in case of MD) and forces for each output frame.
The em.edr file contains information on different energy terms and,
if applicable for the simulation, information on the temperature, pressure, volume and some other quantities during the simulation.
You can extract this information with:
gmx energy -f em.edr -o em_Epot.xvg
xmgrace em_Epot.xvg
gmx trjconv -f em.trr -s em.tpr -o em_centered.xtc -ur compact -pbc mol -centergmx trjconv -f em.trr -s em.tpr -o em_centered.pdb -ur compact -pbc mol -center -dump 0
-dump 0 flag creates a pdb file that contains the coordinates of the peptide at time t=0.
Select the protein group for centering and the whole system for output.
The trajectory will be converted into another file format (*.xtc) that has less precision,
but is much tighter compressed, resulting in smaller files.
Now load the system into VMD with:
vmd em_centered.pdb em_centered.xtc
em.gro.
The information on bonds are automatically generated by VMD.
You can optionally delete the coordinates by right clicking on the molecule in the main window and deleteing the single frame.
This will make the molecule dissapear in the display window,
but the information on atom types and bonds is still retained internally in VMD.
This has the advantage that we will not see a jump from the final configuration
(as loaded from em.gro) to the first frame of the trajectory.
The trajectory can be loaded by selecting File -> New Molecule from the VMD main window.
First select the already loaded molecule from the drop down menu.
Then browse to the trajectory file em_centered.xtc.
The filetype will be detected automatically.
Hit the load button.
Exercises
- Have a look at the trajectory in VMD in some representations of your choice.
- Extract some other quantities of your choice from the
*.edrfile and examine them withxmgrace.