Analysis
System check
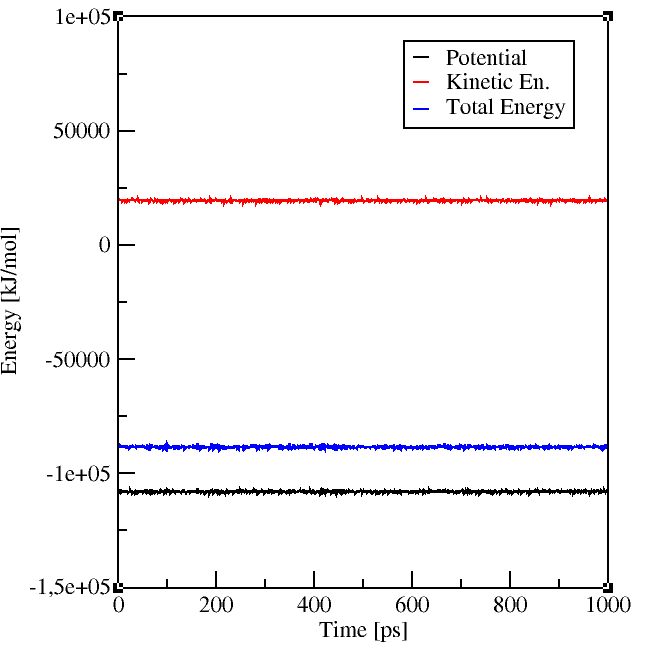
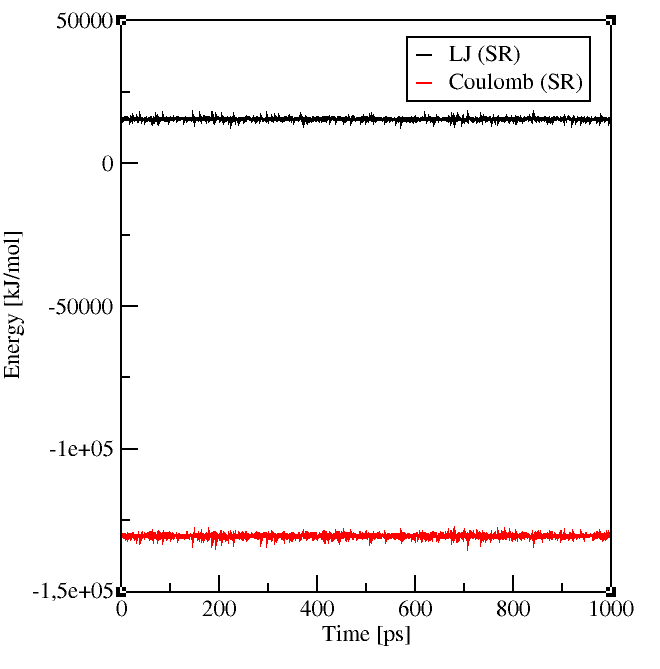
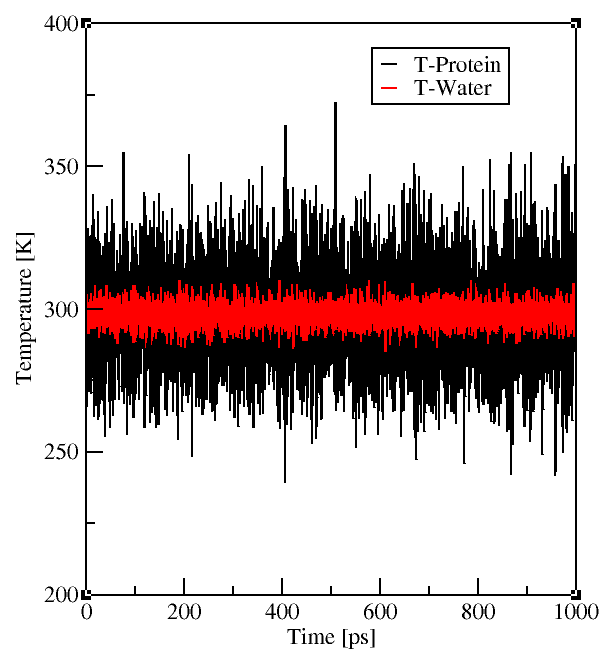
It is also a good idea to look at the energies and pressure of your simulation, best already
during the simulation! If something goes wrong you can realize it before you spent all the precious computing time.
Gromacs enables you to check your system with its energy and traj tools.
gmx energy:
Extracts energy components or distance restraint data from the energy file. The user is prompted to interactively select the energy
terms or other quantities. See all accepted options with gmx energy -h.
gmx energy -f md.edr -o system_term.xvg
gmx trjconv: Can convert trajectory files in many ways. Here, a number of options are chosen to change the
periodicity representation such that the protein is relocated as a whole into the unit cell: gmx trjconv -f md.trr -s md.tpr -o md_protein.trr -center -ur compact -pbc mol
gmx traj: Plots coordinates, velocities, forces and/or the box. With -com the
coordinates, velocities and forces are calculated for the center of mass of each group.
gmx traj -f md_protein.trr -s md_protein.tpr -com -ox coordinates.xvg -ov velocities.xvg -of forces.xvg