Analysis
Clustering
Geometric clustering is performed to identify similar structures sampled during the MD simulation. Various clustering methods exist. One of the most often used clustering algorithms applied to MD trajectories is the one developed by X. Daura (Angew. Chem. Int. Ed.1999, 38, pp 236-240).:
- It is based on the mutual RMSD between all conformations (aka. snapshots) sampled during the MD simulation.
- A RMSD cut-off is chosen to determine cluster membership.
- It counts the number of neighbors for each MD snapshot within the cut-off.
- It identifies the snapshot with the largest number of neighbors (i.e., other snapshots) as cluster center and eleminates it (including all its cluster members) from the pool of snapshots.
- This procedure is repeated for the remaining snapshots in the pool until all snapshots are assigned to a cluster.
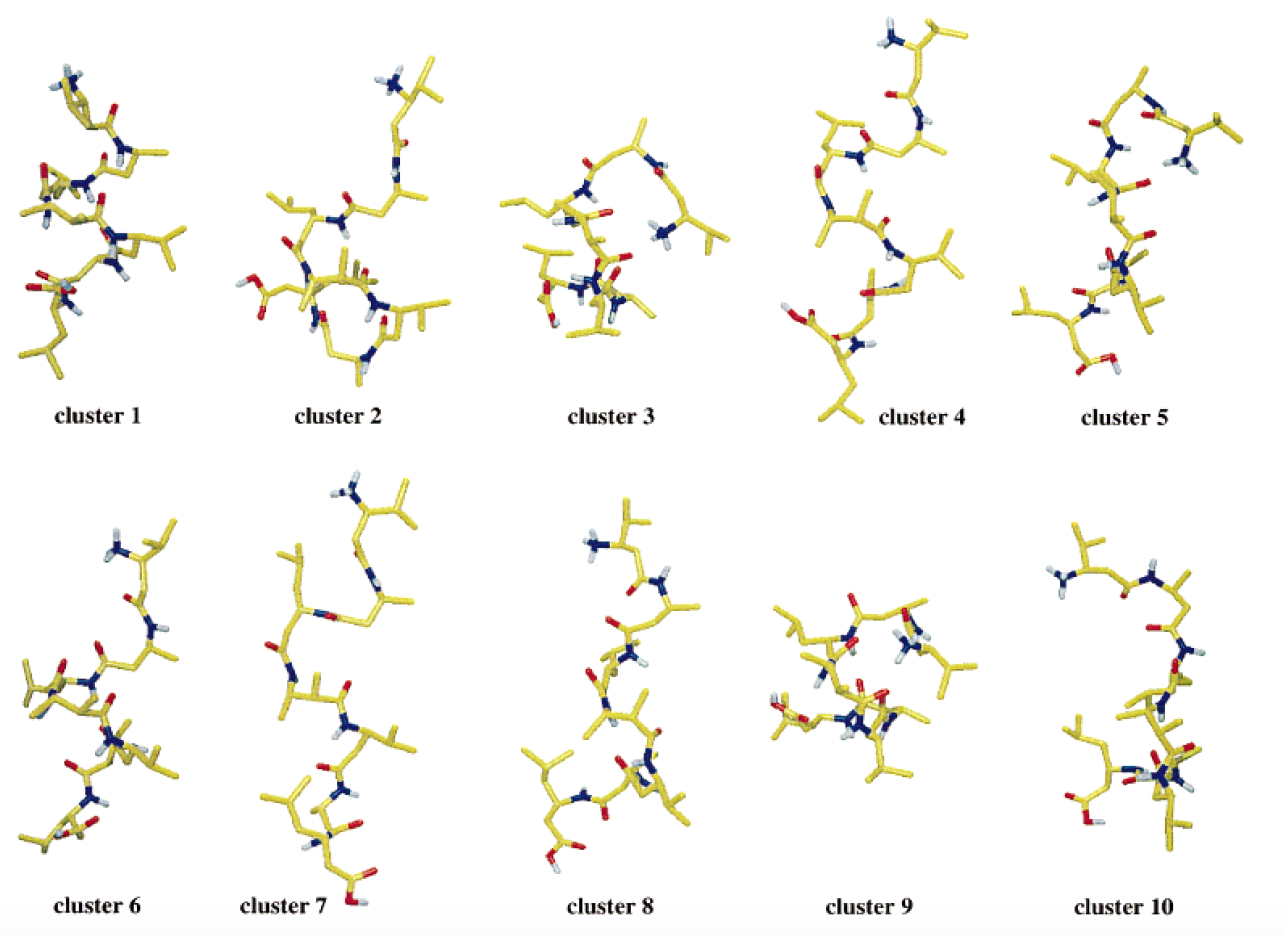
Example: The 10 most populated clusters obtained from a 50-ns MD simulation of a β-heptapeptide in methanol at 340 K; clustering performed with a 0.1 nm cut-off.
gmx cluster can cluster structures with several different methods.
The method option gromos uses the algorithm as described by Daura et al.
gmx cluster -f md_protein.xtc -s md_protein.tpr -method gromos -cl cluster.pdb -g cluster.log -cutoff 0.2
(Selection: "Backbone" for RMSD calculation and "Protein" for output)
cutoff is 0.1 nm, here we use 0.2 nm as structures with an RMSD of 0.1 nm or less
are very similar. The choice of the cut-off also depends on the size and flexibility of the protein under study: the larger and more
flexible a protein is, the larger the cut-off should be chosen.
As the RMSD between all pairs of snapshots of the trajectory has to be calculated, it takes a while until the clustering is
completed. To speed up things, we chose only the "backbone" for the calculation of the RMSD and we could further reduce the time needed via
the flag -dt to reduce the number of snapshots used for the clustering.The midpoint of each cluster is written with the
-cl option, producing the file
cluster.pdb. In addition, information about the number of clusters and their population is written to a log file,
cluster.log.
Use gmx cluster -h
to see all other available options.