Analysis
Hydrogen bonds
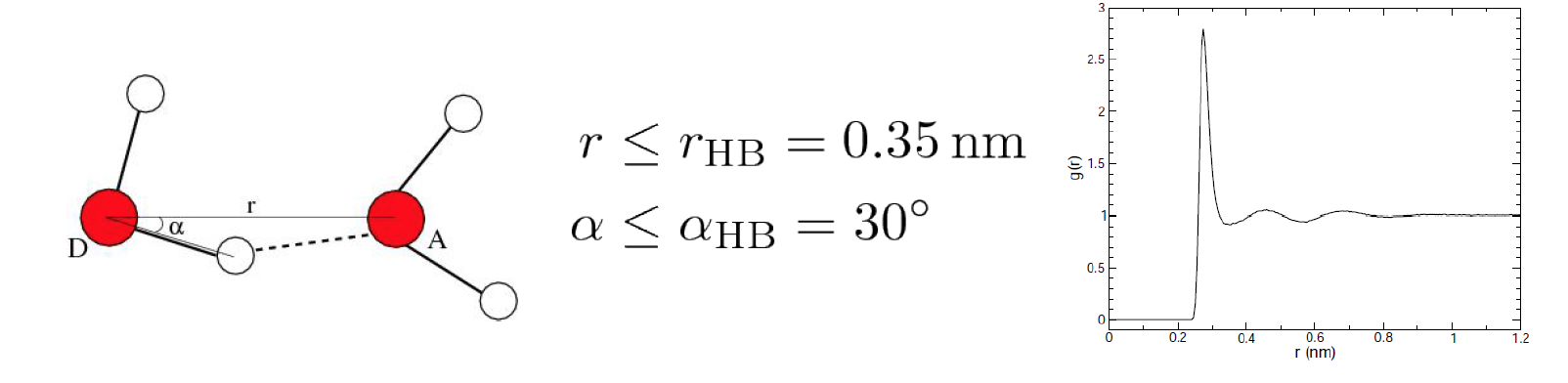

Hydrogen bonds provide most of the directional interactions that underpin protein folding, protein structure and molecular recognition.
The core of most protein structures is composed of secondary structures such as α-helix and β-sheet, which are stabilized by
H-bonds between main chain carbonyl oxygen and amide nitrogen. Hydrogen bonding confers rigidity to the protein structure and
specificity to intermolecular interactions. An H-bond is formed by the interaction of a hydrogen atom that is covalently bonded
to an electronegative atom (donor D) with another electronegative atom (acceptor A). The accepted geometry for
an H-bond is a distance of less than 3.5 Å between hydrogen D and A and an D-H-A angle of
180° ± 30°.
In Gromacs, gmx hbond analyses all H-bonds existing between two groups of atoms
(which must be either identical or non-overlapping)
or in specified D-H-A triplets using specified D-A distance and D-H-A angle criterions. Here, we will perform two commands:
gmx hbond -f md_protein.xtc -s md_protein.tpr -num hbnum_Pro-Pro.xvg -hx hbhelix.xvg
(for intraprotein H-bonds, select two times the "Protein" group)
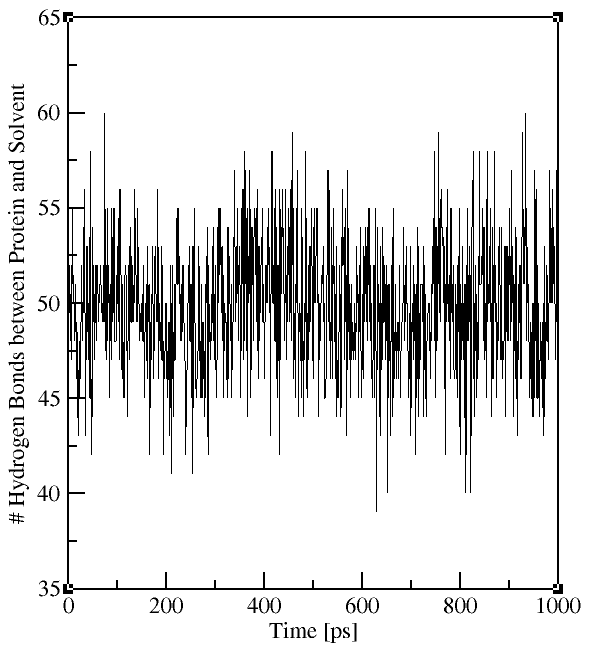
gmx hbond -f md.xtc -s md.tpr -num hbnum_Pro-Sol.xvg
(for H-bonds between protein and water, select "Protein" and "Solvent")
The output file contains the time evolution of the total number
of H-bonds between the selected groups (see plots on the right). The flag -hx provides information on the number of
H-bonds between residues n and n+i with i=0 to 6. The group for i=6 also includes all
H-bonds for i>6. This analysis provides information on the formation of secondary structure, e.g. i=4
for α-helies.